
Transcriptional regulation in human, fly, and worm are conserved
As part of the modENCODE project, the Gerstein lab at Yale did a comprehensive comparison of the transcription of human, fly, and worm. Although separated by millions of years, all three organisms share ancient patterns of gene expression. We built comprehensive models that could be used to predict gene expression across all three organisms. This work was covered in a press release and a News and Views on Nature.
As part of the modENCODE project, the Gerstein lab at Yale did a comprehensive comparison of the transcription of human, fly, and worm. Although separated by millions of years, all three organisms share ancient patterns of gene expression. We built comprehensive models that could be used to predict gene expression across all three organisms. This work was covered in a press release and a News and Views on Nature.
Less is More
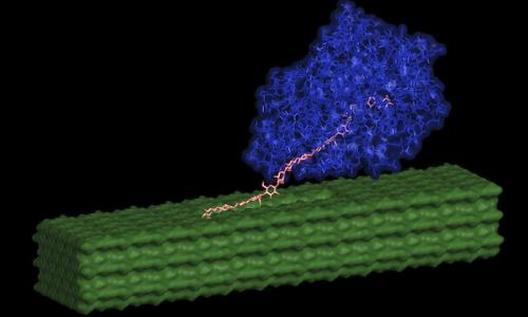
Cellulases decompose cellulose and the production of more efficient cellulase will lead to the production of cheaper biofuels. However, cellulose isolated from plants is crystalline and the efficiency of cellulase depends on the structure of these cellulose crystals and the ease with which different layers of cellulose chain separate from each other. A number of industrial processes have been developed that change the structure of these cellulose crystals and can affect the efficiency of the cellulase. I developed a mechanistic kinetic model for processive cellulases and showed that increased binding of cellulase to cellulose does not always lead to more efficient enzymes. This article was published in the June 2013 issue of the Proceedings of National Academy of Sciences and was highlighted in several websites including Science Daily.
Divide and Conquer
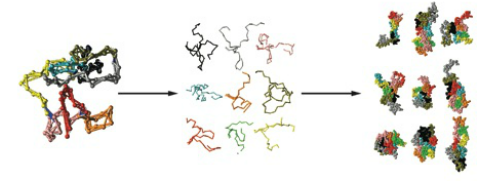
Intrinsically disordered proteins do not have any stable structure and can be found in an ensemble of conformations. Due to the heterogeneous nature of their conformational landscape, they are difficult to characterize using experiments or theory. In collaboration with Gnanakaran, I developed a network analysis method that was utilized in combination with all-atom molecular dynamics simulations to split an intrinsically disordered protein into modular peptides that can be studied in isolation of each other. This article was published and chosen as a research highlight in the August 2012 issue of Biophysical journal. It was also chosen as one of the research highlights in the December 2012 issue of Biopolymers journal.
Quantifying Intramolecular Binding
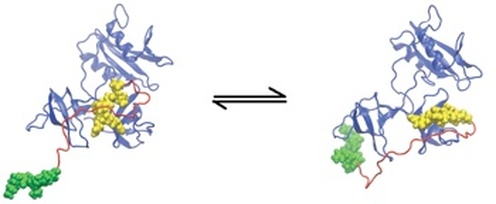
Many biochemical interactions are mediated by multivalent binding in which a number of relatively weak promiscuous interactions to increase the specificity and affinity of complex formation. In collaboration with Gnanakaran and Byron Goldstein, I utilized a combination of bioinformatic, polymer models, molecular dynamics simulations, and thermodynamic modeling to quantify the overall affinity of two multivalent proteins.This article was published in the October 2011 issue of PLoS Computational Biology. These methods can potentially be applied in the design of new biomolecules and drugs.
Signaling Pathways that Sets the Genetic Code

The genetic code is set by a group of enzymes called the aminoacyl-tRNA synthetases. These enzymes recognize the correct pair of amino acid and tRNA molecules from a large number of incorrect combinations. The regions that determine whether the tRNA molecule is correct can be up to 70 Angstroms away from the site of chemistry in these enzymes. In collaboration with Zan Schulten and John Eargle, I developed a network analysis method to determine the most optimal and suboptimal pathways for communication between these regions in these enzymes. The method was published in the April 2009 issue of the Proceedings of National Academy of Sciences, USA. This article was highlightedin prestigious websites like Science Daily.
The evolution of charged tRNA formation
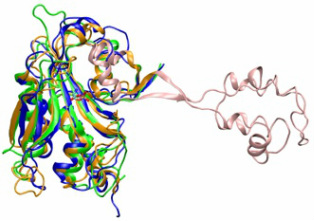
|
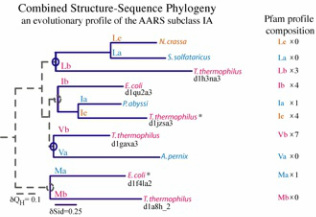
The mechanism for cysteine aminoacylation remained unknown in a number of methanogens a decade after the first methanogenic genome was sequenced. During my doctoral research, I developed a new bioinformatic tool that combined sequence and structural information to perform the evolutionary analysis of biomolecular families (top figure). Using this approach, I was able to identify the gene involved in cysteine aminoacylation in methanogens (bottom figure). Dieter Soll and coworkers performed elegant biochemical experiments and proved that this gene was indeed involved in tRNA aminoacylation and was also important for amino acid biosynthesis in methanogens. Even though this gene is only present in most methanogens, we showed that this gene was present in the last univeral common ancestor(s). The bioinformatic tool and the corresponding evolutionary study were published in the March and December(2005) issues of the Proceedings of National Academy of Sciences. This article was highlighted in several science news sites including Science Daily.
|
